Visium空间转录组分析流程
# Analysis, visualization, and integration of spatial datasets with Seurat at https://satijalab.org/seurat/articles/spatial_vignette.html
# set path ------------------------------------------
rm(list=ls());options(stringsAsFactors=FALSE)
project_dir <- rprojroot::find_rstudio_root_file()
raw_dir <- paste0(project_dir,"/raw-data/")
process_dir <- paste0(project_dir,"/process-data/")
output_dir <- paste0(project_dir,"/output-results/")
temp_dir <- paste0(project_dir,"/temp-data/")
bin_dir <- paste0(project_dir,"/bin/")
script_dir <- paste0(project_dir,"/workflow-scripts/")
work_dir <- output_dir
setwd(work_dir)
# library and set default parameter ----------------
library(tidyverse)
library(Seurat)
library(SeuratData) #devtools::install_github('satijalab/seurat-data')
library(patchwork)
library(edgeR)
library(SingleCellExperiment)
prefix <- "Seurat-3-"
output_prefix <- paste0(output_dir,prefix)
创建一些函数
# functions -------------
run_clustering <- function(data, resolution = 0.1, npcs=30) {
data <- SCTransform(data, assay = "Spatial", verbose = TRUE)
data <- RunPCA(data, assay = "SCT", verbose = TRUE)
data <- FindNeighbors(data, reduction = "pca", dims = 1:npcs)
data <- FindClusters(data, verbose = TRUE, resolution=resolution)
data <- RunUMAP(data, dims = 1:npcs)
return(data)
}
pooling_seu <- function(seu_obj,assay="SCT") {
cell_metadata <- seu_obj@meta.data
# create sudo id by random sample into 3 sets from each patients
cell_metadata$sudo_sample_id <- NA
samples <- unique(cell_metadata$orig.ident)
nrg <- 3
for(i in c(1:length(samples))){
set.seed(i)
temp_sample_idx <- which(cell_metadata$orig.ident==samples[i])
sample_sudo_idx <- sample(c(1:nrg),length(temp_sample_idx),replace=T,prob=rep(1/nrg,nrg)) # random sample into 3 sets
cell_metadata[temp_sample_idx,'sudo_sample_id'] <- paste(samples[i],sample_sudo_idx,sep='_')
}
sce <- SingleCellExperiment(assays = list(counts = GetAssayData(seu_obj,slot="count",assay=assay)), colData = cell_metadata)
summed <- scuttle::aggregateAcrossCells(sce, id=colData(sce)[,'sudo_sample_id']) # aggregate counts by id
return(summed)
}
dga_treat <- function(DGEList,ref_level=NA,set_regulate=TRUE){
DGEList$samples$lib.size <- colSums(DGEList$counts)
if(is.na(ref_level)){
DGEList$samples$group <- relevel(DGEList$samples$group, ref = levels(DGEList$samples$group)[1] )
} else{
DGEList$samples$group <- relevel(DGEList$samples$group, ref = ref_level)
}
keep <- filterByExpr(DGEList, group=DGEList$samples$group) #set your own threshold
table(keep)
DGEList <- DGEList[keep,, keep.lib.sizes=FALSE]
DGEList <- calcNormFactors(DGEList)
# DE
design <- model.matrix(~DGEList$samples$group)
DGEList <- estimateDisp(DGEList, design, robust=TRUE)
fit <- glmQLFit(DGEList, design, robust=TRUE)
#lrt <- glmQLFTest(fit, coef=ncol(fit$design))
lrt <- glmTreat(fit, coef=ncol(fit$design), lfc=log2(1.2))
print(table(decideTests(lrt)))
res <- topTags(lrt,n = nrow(DGEList))
if(set_regulate){
res$table$regulate <- "Normal"
res$table$regulate[res$table$logFC>0 & res$table$FDR<0.05] <- "Up"
res$table$regulate[res$table$logFC<0 & res$table$FDR<0.05] <- "Down"
res$table <- res$table[order(res$table$FDR),]
}
return(res)
}
plot_rctd <- function(RCTD,seu_obj, image_name="VLP43_kidney_A1",r=3){
stopifnot("RCTD"%in% class(RCTD))
stopifnot("FindClusters"%in% names(seu_obj@commands))
norm_weights <- normalize_weights(RCTD@results$weights) # normalize the cell type proportions to sum to 1.
query <- subset(seu_obj,cells = row.names(RCTD@spatialRNA@coords))
pos <- RCTD@spatialRNA@coords
annot <- query$seurat_clusters
p <- vizAllTopics(as.matrix(norm_weights), pos,
#topicCols = qualitative_hcl(n=ncol(norm_weights),palette="Dynamic"),
topicCols = brewer.pal(n=ncol(norm_weights),name = "Paired"),
groups = annot,
group_cols = rainbow(length(levels(annot))),
#group_cols = rainbow_hcl(length(levels(annot))),
r=r,lwd = 0.1) + labs(title=image_name) +
#ggplot2::guides(colour = "none") +
theme(plot.background = ggplot2::element_rect(fill = "white"),plot.title = element_text(hjust = 0.5,size=15))
return(p)
}
Visium空间转录组技术
当前时代的空间转录组技术大致分为五类方向:激光显微切割(laser capture microdissection,LCM),单分子荧光原位杂交(single molecular fluorescent in situ hybridization, smFISH),靶向原位测序(In situ sequencing,ISS),原位阵列捕获(In situ array capture, Array) 和 其他非成像技术(No imaging)。
目前商业用的最广的还是Visium平台,因此我们以Visium数据为例进行演示,进行如聚类,差异表达,反卷积等分析。使用的工具是Seurat,spacexr等包。在单细胞下游分析中,Seurat是一个非常强大的R包,不仅有强大的社区、详细文档说明还有很多内置数据。这里以Analysis, visualization, and integration of spatial datasets with Seurat 数据为例进行空间转录组分析。
1. 数据探索
查看测序数据质量,是否符合生物学背景。如果质量不好,需要进行质量控制。stxBrain已经是过滤后的数据,我们不需要进行质量控制。
#InstallData("stxBrain") # 如果由于网络问题,安装失败。可以将url放在浏览器中下载tar.gz文件,然后本地安装。
brain <- LoadData("stxBrain", type = "anterior1")
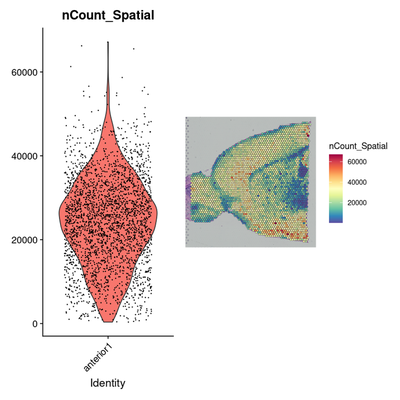
# Data preprocessing: Normalization
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial") + theme(legend.position = "right")
wrap_plots(plot1, plot2)
# 斑点上分子计数的变化不仅是技术上的问题,而且还取决于组织的解剖结构。
# 例如,神经元(例如皮质白质)耗竭的组织区域可再现地显示出较低的分子数。
2. 聚类
和单细胞数据类似,我们需要对数据进行聚类。也可以使用run_clustering函数。
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
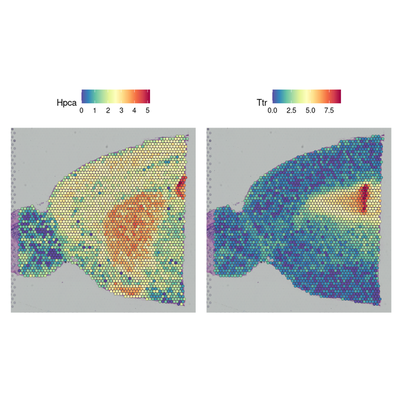
# Gene expression visualization
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 1)
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
p1 + p2
# Dimensionality reduction, clustering
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
# visualization
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
p1 + p2
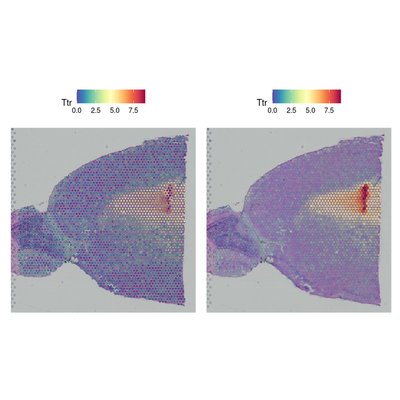
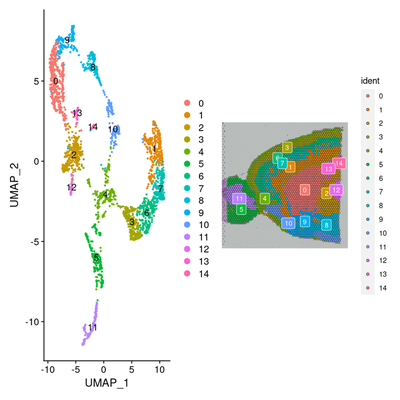
3. 检测亚群特异和空间特异差异基因
对不同亚群进行差异分析,对不同空间分布中的细胞进行基因差异分析。
# Detecting spatially-variable features
de_markers <- FindMarkers(brain, ident.1 = 5, ident.2 = 6)
head(de_markers)
SpatialFeaturePlot(object = brain, features = rownames(de_markers)[1:3], alpha = c(0.1, 1), ncol = 3)
| p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | |
|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | |
| Calb2 | 6.427214e-69 | 3.336874 | 1.000 | 0.537 | 1.135560e-64 |
| Camk2n1 | 1.519204e-68 | -2.450388 | 1.000 | 1.000 | 2.684130e-64 |
| Nrgn | 1.573095e-68 | -3.229826 | 0.971 | 1.000 | 2.779344e-64 |
| Stx1a | 2.104119e-68 | -2.238353 | 0.784 | 1.000 | 3.717558e-64 |
| Nptxr | 9.820482e-68 | -1.918293 | 0.942 | 1.000 | 1.735083e-63 |
| Lingo1 | 2.124777e-67 | -1.913281 | 0.880 | 1.000 | 3.754056e-63 |
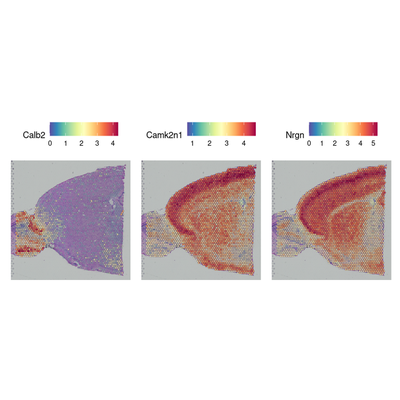
#在没有预注释的情况下搜索表现出空间模式的特征
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT", features = VariableFeatures(brain)[1:1000],selection.method = "moransi") # moransi mothed, markvariogram too slow
#top.features <- head(SpatiallyVariableFeatures(brain, selection.method = "moransi"), 6)
top.features <- c("Calb2","Gng4","Ttr","S100a5","Nrgn","Doc2g")
SpatialFeaturePlot(brain, features = top.features, ncol = 3, alpha = c(0.1, 1))
Computing Moran's I
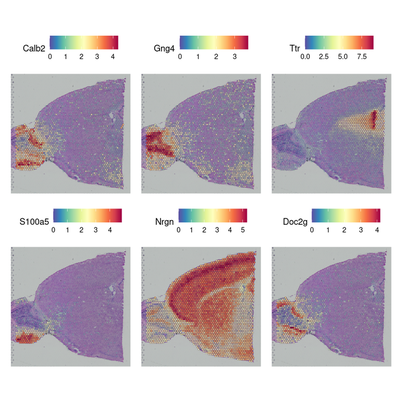
4. 检测分组特异差异基因
由于此数据没有分组信息,我们导入另一个数据进行演示。由于直接对分组细胞进行差异分析会造成假阳性,这里我们用psudo-bulk差异分析
brain <- readRDS("/media/zhangfeng/myData/projects/sp-aging/output-results/70-visium-brain-cluster.RData")
# DEG group--------
table(brain@meta.data$group)
summed <- pooling_seu(brain)
class(summed)
head(summed@colData)
is_ribo<-grepl('Rps|Rpl',row.names(summed))
is_mito<-grepl('mt-',row.names(summed))
y <- DGEList(counts(summed)[(!is_ribo)&(!is_mito),], samples=colData(summed)$sudo_sample_id,group=colData(summed)$group) # DE between types
res <- dga_treat(DGEList=y,ref_level = "young")
head(res$table)
aged young 10715 11479
DataFrame with 6 rows and 11 columns
orig.ident nCount_Spatial nFeature_Spatial group nCount_SCT
<character> <numeric> <integer> <character> <numeric>
VLP40_1A_1 VLP40_1A NA NA young NA
VLP40_1A_2 VLP40_1A NA NA young NA
VLP40_1A_3 VLP40_1A NA NA young NA
VLP40_1B_1 VLP40_1B NA NA young NA
VLP40_1B_2 VLP40_1B NA NA young NA
VLP40_1B_3 VLP40_1B NA NA young NA
nFeature_SCT SCT_snn_res.0.1 seurat_clusters sudo_sample_id
<integer> <factor> <factor> <character>
VLP40_1A_1 NA NA NA VLP40_1A_1
VLP40_1A_2 NA NA NA VLP40_1A_2
VLP40_1A_3 NA NA NA VLP40_1A_3
VLP40_1B_1 NA NA NA VLP40_1B_1
VLP40_1B_2 NA NA NA VLP40_1B_2
VLP40_1B_3 NA NA NA VLP40_1B_3
ids ncells
<character> <integer>
VLP40_1A_1 VLP40_1A_1 1023
VLP40_1A_2 VLP40_1A_2 1041
VLP40_1A_3 VLP40_1A_3 1025
VLP40_1B_1 VLP40_1B_1 915
VLP40_1B_2 VLP40_1B_2 915
VLP40_1B_3 VLP40_1B_3 865
-1 0 1
87 14105 203
| logFC | unshrunk.logFC | logCPM | PValue | FDR | regulate | |
|---|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <chr> | |
| Lyz2 | 2.6841911 | 2.6854073 | 5.326010 | 2.446786e-17 | 2.992338e-13 | Up |
| C4b | 3.0082676 | 3.0088154 | 6.819937 | 5.386843e-17 | 2.992338e-13 | Up |
| Itgax | 3.8597675 | 3.9129263 | 1.172032 | 6.236203e-17 | 2.992338e-13 | Up |
| Cd52 | 1.3891682 | 1.3899204 | 4.541512 | 1.229941e-15 | 4.426248e-12 | Up |
| Pcdhb9 | 0.9295853 | 0.9301349 | 4.305468 | 2.580643e-15 | 7.429671e-12 | Up |
| Fcgr2b | 1.4622417 | 1.4650595 | 2.751271 | 1.811064e-14 | 4.032808e-11 | Up |
5. Deconvolution分析
由于Visium平台的spot不是单细胞分辨率,一般有1-10个细胞,因此我们需要对其进行注释,分析不同细胞的比例。这里使用spacexr包进行分析。
reference <- readRDS('/media/zhangfeng/myData/projects/sp-aging/output-results/4-brain-SCRef.rds')
# control sample
spatial_controls <- RCTD_replicates(seu_obj = brain[,brain$group=="young"])
RCTD_controls <- create.RCTD.replicates(spatial_controls$spatialRNA_replicates, reference, spatial_controls$samples, max_cores = 20)
RCTD_controls <- run.RCTD.replicates(RCTD_controls, doublet_mode = 'full')
# treatment samples
spatial_treats <- RCTD_replicates(seu_obj = brain[,brain$group=="aged"])
RCTD_treats <- create.RCTD.replicates(spatial_treats$spatialRNA_replicates, reference, spatial_treats$samples, max_cores = 20)
RCTD_treats <- run.RCTD.replicates(RCTD_treats, doublet_mode = 'full')
# add into seurat
spot_weight <- combine_weight(RCTD_controls,RCTD_treats)
meta_data <- brain@meta.data %>% rownames_to_column("spot_id") %>%
left_join(spot_weight,by="spot_id")
all(meta_data$spot_id==colnames(brain))
meta_data <- column_to_rownames(meta_data,"spot_id")
brain <- AddMetaData(brain,metadata = meta_data)
saveRDS(brain,file = "/media/zhangfeng/myData/projects/sp-aging/output-results/70-visium-brain-RCTD.RData"))
brain <- readRDS("/media/zhangfeng/myData/projects/sp-aging/output-results/70-visium-brain-RCTD.RData")
head(brain@meta.data)
| orig.ident | nCount_Spatial | nFeature_Spatial | group | nCount_SCT | nFeature_SCT | SCT_snn_res.0.1 | seurat_clusters | Astrocytes | Ependymal | Immune | Microglia | Neurons | Oligos | PeripheralGlia | Vascular | max_cell | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <dbl> | <int> | <chr> | <dbl> | <int> | <fct> | <fct> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <chr> | |
| AAACAAGTATCTCCCA-1_1_1_1 | VLP40_1A | 8421 | 3160 | young | 15335 | 3514 | 3 | 3 | 0.19143611 | 0.0006158916 | 0.0049827905 | 0.004197530 | 0.3236210 | 0.433052200 | 0.011450442 | 0.030644049 | Oligos |
| AAACACCAATAACTGC-1_1_1_1 | VLP40_1A | 23006 | 5649 | young | 16862 | 5507 | 0 | 0 | 0.11293516 | 0.0013890113 | 0.0006617161 | 0.011798306 | 0.8054124 | 0.062050025 | 0.002048477 | 0.003704872 | Neurons |
| AAACAGAGCGACTCCT-1_1_1_1 | VLP40_1A | 25131 | 6224 | young | 16782 | 5926 | 0 | 0 | 0.04871926 | 0.0073010556 | 0.0026329875 | 0.009357506 | 0.9132606 | 0.004827619 | 0.002313284 | 0.011587694 | Neurons |
| AAACAGCTTTCAGAAG-1_1_1_1 | VLP40_1A | 35993 | 7074 | young | 16298 | 5253 | 0 | 0 | 0.05981760 | 0.0019247194 | 0.0001026660 | 0.013729242 | 0.8918254 | 0.007285013 | 0.002966418 | 0.022348983 | Neurons |
| AAACAGGGTCTATATT-1_1_1_1 | VLP40_1A | 37738 | 7220 | young | 16163 | 5196 | 0 | 0 | 0.05777206 | 0.0014648800 | 0.0009153018 | 0.007876570 | 0.9005069 | 0.023966030 | 0.000675534 | 0.006822758 | Neurons |
| AAACATTTCCCGGATT-1_1_1_1 | VLP40_1A | 14901 | 4544 | young | 15460 | 4544 | 3 | 3 | 0.13274107 | 0.0026031003 | 0.0001558520 | 0.006728277 | 0.6066684 | 0.218612193 | 0.010717393 | 0.021773681 | Neurons |
6. 绘图
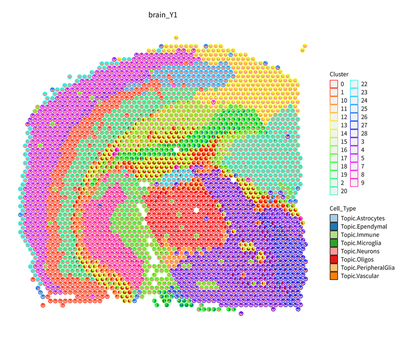
slice_id <- names(RCTD_controls@group_ids)[1]
myRCTD_full <- RCTD_controls@RCTD.reps[[1]]
p_brain <- plot_rctd(myRCTD_full,brain,image_name = slice_id)+ggtitle(slice_id)+
theme(plot.title=element_text(hjust = 0.5))+labs(fill="Cell_Type",color="Cluster")+
guides(colour = "none")
p_brain